
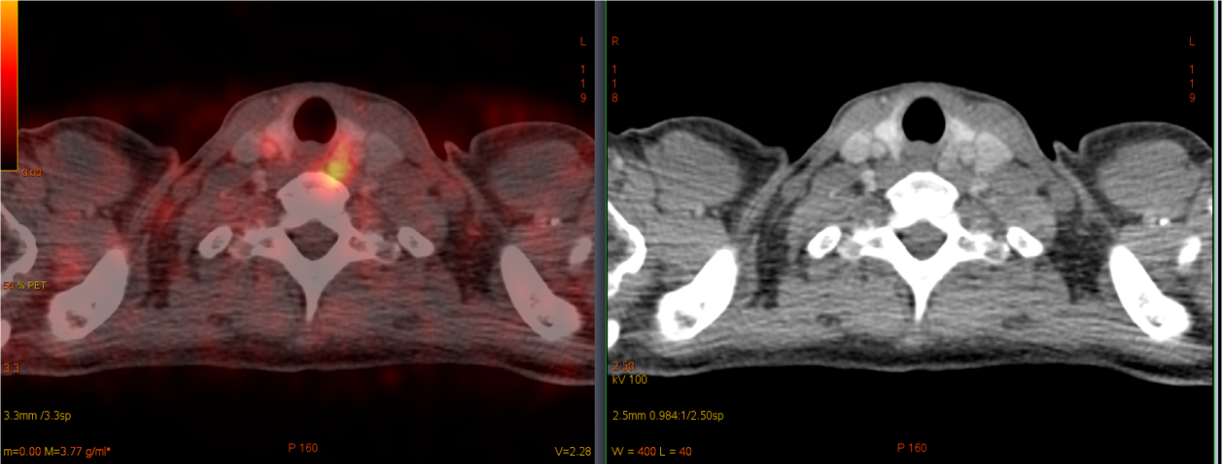
68Ga-DOTA-TATE PET/CT: a focus of radiopharmaceutical accumulation in the area of the upper left thyroid lobe (Fig. 1), consistent with the lesion on CT scan (Fig. 2).
Scientists of the WCRC for Personalized Medicine discovered new possibilities for the treatment of parathyroid adenoma and primary hyperparathyroidism in patients with multiple endocrine neoplasia type 1.
Multiple endocrine neoplasia type 1 (MEN 1) is a rare hereditary tumor syndrome caused by mutation in the gene encoding a tumor suppressor protein, menin. Normally, this protein prevents tumor growth. Major manifestations of this syndrome are neuroendocrine tumors of various sites. The most common tumors are those of the parathyroid glands, pituitary gland, and pancreas. Parathyroid damage in this genetic syndrome is that adenomas develop in all four glands. Therefore, according to modern guidelines, it is necessary to remove 3.5 or 4 parathyroid glands. However, this approach often leads to the development of hypoparathyroidism (decreased functions of the parathyroid glands after surgery), a serious disorder with limited possibilities for adequate treatment. For this reason, total removal is rarely performed and is not always technically possible.
The search for methods of medical therapy for parathyroid adenomas that develop in MEN 1 syndrome remains an urgent challenge. It is not uncommon for patients with MEN 1 to develop multiple neuroendocrine tumors of the pancreas and gastrointestinal tract that require medical treatment after surgery. The drugs of choice in these cases are first-generation somatostatin analogues.
To visually observe drug sensitivity, doctors use positron emission tomography combined with computed tomography (PET / CT) with radiopharmaceuticals. In PET / CT, cells with somatostatin receptors on their surface «glow» in the image.
Researchers at the Research Laboratory of Neuroendocrine Tumors of the WCRC for Personalized Medicine for the first time obtained data on the possibility of using somatostatin analogues in MEN 1 patients for the treatment of primary hyperparathyroidism that persists after removal of three parathyroid adenomas.
A patient with neuroendocrine tumors of the pancreas, stomach and duodenum had a PET/CT scan. The scan revealed an increased accumulation of a radiopharmaceutical by pancreatic neuroendocrine tumors and a focus of radiopharmaceutical accumulation in the area of the lower pole of the left thyroid lobe. Previous ultrasound, CT and scintigraphy proved that this lesion is a parathyroid adenoma. Thus, data were obtained on the expression of type 2 somatostatin receptors in parathyroid adenoma in a patient with MEN 1 syndrome.
The patient underwent surgical treatment for tumors of the pancreas, stomach and duodenum. After the surgery, therapy with first-generation somatostatin analogues was prescribed. The levels of total and ionized calcium returned to normal, and the level of parathyroid hormone decreased significantly. Thus, the use of a somatostatin analogue in a patient with MEN 1 syndrome made it possible to avoid parathyroid surgery and postoperative hypoparathyroidism.
The results obtained by WCRC researchers open up new prospects in the treatment of this difficult group of patients. Further research will expand the understanding of the possibilities of personalized medical therapy for parathyroid neuroendocrine tumors in the MEN 1 syndrome.